Highlights
These researchers visualized the spike proteins of SARS-CoV-2 and the most closely related bat virus RaTG13 and compared the structure and function of the two proteins. The images of the SARS-CoV-2 spike protein the researchers produced allowed them to reconstruct the 3D model of the spike protein at the highest resolution available to date. The findings show that SARS-CoV-2 could not have come directly from the bat viruses we know, because the bat coronavirus does not bind to the human receptor like SARS-CoV-2 does. Rather, there must have been another animal intermediary or another bat precursor virus that we still have not found, possibly the Malayan pangolin, Manis javanica.
All of our lives have been transformed by the outbreak of
COVID-19
in the U.S. and around the world. Since COVID-19 was first reported in China, people across the globe have been wondering: where did COVID-19 come from? Ongoing research by Dr. Antoni Wrobel and Dr. Donald Benton, postdoctoral fellows in the laboratory of Dr. Steve Gamblin at the Francis Crick Institute in London, UK, seek to answer this question by comparing the virus that caused COVID-19 in humans to similar viruses in other animals.
Although many of us had never heard the word
coronavirus
before late last year, coronaviruses have most likely been with humans for centuries. Typically, coronaviruses in humans cause seasonal respiratory illnesses that we think of as the common cold. In animals, coronaviruses can cause
gastrointestinal
and respiratory symptoms. The family of coronaviruses is quite large and can be classified into four groups: alpha, beta, gamma, and delta. Only alpha and beta coronaviruses are known to infect humans.
Coronavirus was first discovered in humans in the 1960s. When examining the virus under an electron microscope, researchers noticed decorative spikes all around it that reminded them of the halo around the sun during a
solar eclipse,
known as a solar corona. As a result, they named the new virus coronavirus.

Figure 1. Solar corona during total solar eclipse
[Source: https://spaceplace.nasa.gov/sun-corona/en/]
Compared to other viruses, coronaviruses have much more genetic material, which can mutate to adapt to new
hosts.
Interestingly, when coronaviruses encounter other different coronaviruses, they can swap fragments of their genetic material, a phenomenon that scientists call genetic
recombination.
This is one reason why outbreaks of infections like
Middle Eastern Respiratory Syndrome
(MERS) in 2012 and
Severe Acute Respiratory Syndrome
(SARS) in 2003 were caused by coronaviruses. This is also the reason why outbreaks of coronavirus-related epidemics in humans tend to occur quite often, about every 10-20 years. This is how often different animal coronaviruses can mutate and recombine to form new viruses capable of infecting humans.
There is still much scientific debate about where exactly
SARS-CoV-2,
the novel coronavirus that causes COVID-19, came from. There is consensus in the scientific community that the “ancestors” of the virus somehow had to come from bats, because SARS-CoV-2 belongs to the subgenus Sarbecovirus of beta-coronaviruses that mostly infect bats. However, it remains unknown whether it came directly from bats and when the “ancestor” of SARS-CoV-2 moved from bats to humans. One hypothesis is that the virus or its “ancestor” jumped species from bats directly to humans. A competing hypothesis is that the virus mutated and recombined its way from bats to humans through an intermediary species, possibly the
pangolin,
a nocturnal mammal also known as a scaly anteater.

Figure 2. Malayan pangolin Manis javanica
[Source: https://en.wikipedia.org/wiki/Sunda_pangolin]
To better understand the origin of SARS-CoV-2, Dr. Wrobel, Dr. Benton, and colleagues in the Structural Biology of Disease Processes Laboratory under the supervision of Dr. Gamblin analyzed proteins of the SARS-CoV-2 and compared them to those of the most similar bat coronavirus RaTG13.
In particular, Dr. Wrobel and his colleagues focused on the proteins responsible for the characteristic spiky appearance of the coronaviruses. These proteins, called “spike proteins”, “S proteins” or simply “spikes” for short, are
transmembrane proteins.
This means that part of the protein is located outside the virus, a part remains embedded inside the membrane, and a part is located inside the virus. For the purposes of these experiments, Dr. Wrobel and his colleagues focused only on the largest part of the S protein located outside the virus. This is because it is the external part of the S protein that facilitates infection by attaching the virus to host
receptor proteins.
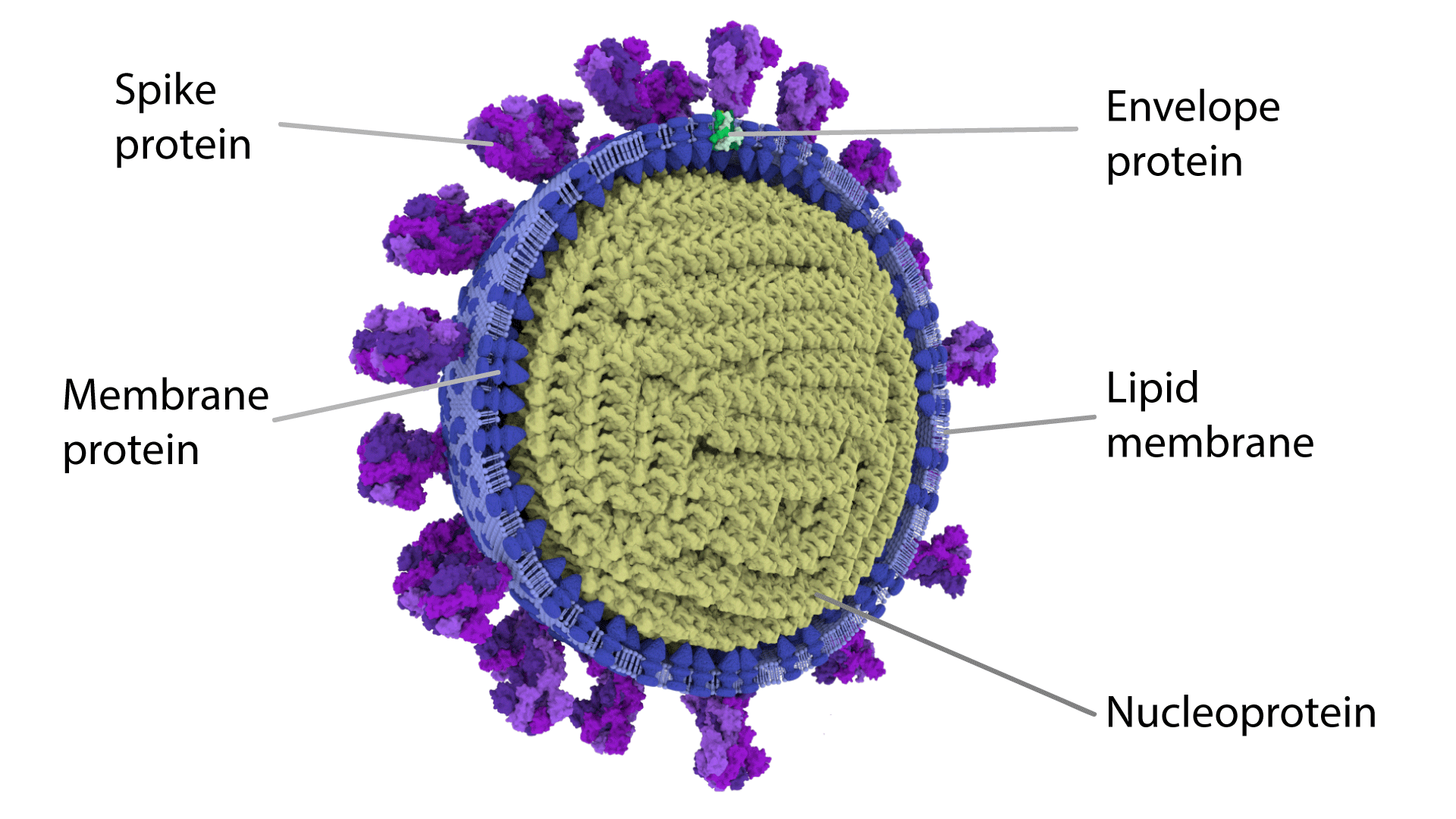
Figure 3. Structure of coronavirus including the spike (S) protein
[Source: https://coronavirusexplained.ukri.org/images/article/Half-virion.png]
The receptor protein that coronaviruses bind to in human cells is called angiotensin-converting enzyme 2 or ACE2 for short. In order for infection to take place, the SARS-CoV-2 protein must bind to the ACE2 receptor.
Prior to the COVID-19 outbreak, Dr. Wrobel, Dr. Benton and their colleagues specialized in the study of proteins that cause disease. One particular area of research was influenza viruses, which can cause the “common” flu, as well as more deadly diseases such as the 1918 pandemic known as the
“Spanish Flu.”
The researchers analyzed the structure of proteins in the flu virus to understand how they work and to track how new
virus strains
evolve and become capable of infecting new hosts. As it became clear that COVID-19 was not going away, Drs. Wrobel and Benton began a similar project with SARS-CoV-2.
The research conducted by Drs. Wrobel and Benton would not have been possible without the previous contributions of Chinese researchers. In order to analyze a structure of any protein, the first step is to understand its genetic sequence. Several weeks into the COVID-19 outbreak in China, Chinese researchers published the complete viral genome of SARS-CoV-2. Dr. Wrobel and colleagues used this information to construct artificial genes for production of the external part of the SARS-CoV-2 spike protein in the lab.
In addition to the genetic sequence of the SARS-CoV-2 spike, the genetic construct also had an additional piece of genetic information that is translated into a small tail attached to the end of the protein. This tail allows the protein to bind to something that it normally does not bind to, in this case small beads decorated with cobalt ions. This allows the researchers to extract the target protein more easily from the rest of the cellular matter because, in the entire mixture, only the target proteins have the tail that can easily bind to cobalt. Once the protein was extracted, the cobalt beads were removed, and the protein was further purified to remove any remaining cellular debris.
In the laboratory, Dr. Wrobel used the constructed genes to produce the external part of the SARS-CoV-2 spike protein. This was done by introducing the genetic information that encodes the SARS-CoV-2 spike construct described above into mammalian cells, which then produced the corresponding protein. In order to extract the protein produced by mammalian cells from the cellular medium, Dr. Wrobel added the cobalt beads, to extract and purify the protein, as described in the paragraph above.
The next step was to visualize the purified spike protein to better understand its structure. For this, Dr. Wrobel and Dr. Benton used
cryogenic electron microscopy
or Cryo EM. Cryo EM is a technique where the sample is frozen before analysis with an electron microscope. The electron microscope is so powerful that it can allow the researchers to see individual bonds between atoms.

Figure 4. Dr. Benton (left) and Dr. Wrobel (right) in front of the cryogenic electron microscope
[Source: Image courtesy of Dr. Wrobel]
Dr. Wrobel and Dr. Benton froze the samples with liquid ethane rather than a regular freezer. This is important because the protein molecules are dissolved in a water solution. If the solution were to be frozen in a regular freezer, the water in the solution would create ice crystals, which would then destroy the proteins. You can see how this works at home by comparing the stiffness of a fresh cucumber to one that has been frozen and thawed. The reason the frozen cucumber is unpleasant and limp is because of the damage the ice did to the cucumber cells. Samples frozen in a regular freezer would be about as useful as a frozen cucumber.
To avoid this problem, Dr. Wrobel and Dr. Benton froze the samples using liquid ethane. The researchers took single droplets of the protein solution and placed them on a small copper disc also referred to as a grid. They blotted the sample between filter papers in order to leave only a very thin layer of the solution on the grid, and then plunged it into liquid ethane. Ice that forms this way is called vitreous ice, which is ice without ice crystals that, to some extent, resembles clear glass.

Figure 5. Single droplets of the protein solution are placed on small copper discs for freezing
[Source: Image courtesy of Dr. Wrobel]
Dr. Wrobel and Dr. Benton used the frozen samples to visualize the protein structure under the electron microscope. Since microscope images are two-dimensional representations of three-dimensional proteins, each single protein image is only one view of the whole protein. For this reason, Dr. Wrobel and Dr. Benton had to collect many images with different views to piece together the overall 3D protein structure.

Figure 6. Picture of Vitrobot that freezes the copper discs
[Source: Image courtesy of Dr. Wrobel]
Previous research, including work done by scientists studying other types of coronavirus spike proteins, helped Dr. Gamblin's team piece together the structure of the SARS-CoV-2 spike protein. “We were able to piece together the structure of the spike protein so quickly because of all the research that has come before,” commented Dr. Wrobel. Dr. Wrobel estimates what they were able to do in just a few days might have taken over a year without any previous templates to guide them.
At the time of its publication in July 2020, the
structure
of the SARS-CoV-2 spike protein obtained by Drs. Wrobel and Benton was the highest resolution and most complete structure available. This is also helpful for other researchers who can use work of Dr. Gamblin's team as a basis for their own ongoing research into SARS-CoV-2 and COVID-19.
Once the team created a three-dimensional structure of the SARS-CoV-2 spike protein from the human virus, the next step was to compare it to the spike protein of the most similar coronavirus in animals. This is a coronavirus found in bats, whose genetic information was released by researchers in China around the same time they released the complete genome of SARS-CoV-2. The spike protein of the bat coronavirus shares about 97% similarity in protein sequence to SARS-CoV-2. Drs. Wrobel and Benton used a similar process to visualize the bat coronavirus with Cryo EM.
They found the two spike proteins looked similar, but they were different in one very important respect. The bat coronavirus was unable to bind to the human receptor, meaning it could not infect humans.
The researchers created the human receptor protein with techniques similar to those used to create the spike proteins. Then they mixed the human receptor protein with a spike protein and measured how strongly the two proteins interacted. For SARS-CoV-2, the spike protein bound quite tightly to the human ACE2 receptor. This was expected, since we know that SARS-CoV-2 can infect humans.
However, the spike protein of the bat RaTG13 coronavirus did not bind significantly to the human receptor protein. This finding indicates this genetically close bat coronavirus could not have just jumped species to infect humans, because the bat virus would not be able to bind to the human receptors that would allow it to cause disease. This means there are some bat viruses that were the ancestors of SARS-CoV-2 that we have yet to discover, or there would have to be one or more intermediaries between the bat coronavirus and SARS-CoV-2.
One of many hypotheses for the evolution of SARS-CoV-2 is that it went from bats to humans via the pangolin. To gain further insights into pangolin viruses, Drs. Wrobel and Benton have been conducting experiments using the spike proteins of the coronaviruses recently found in pangolins. Pangolins are one of the most trafficked wild animals in Asia due to their use in traditional Chinese medicine.
According to one hypothesis, a coronavirus from bats was transferred to pangolins and then jumped from pangolins to humans, possibly via a human-pangolin interaction during wildlife trafficking. Drs. Wrobel and Benton have recently tested this hypothesis in their latest work comparing the SARS-CoV-2 virus with its closest bat coronavirus RaTG13 and its closely related pangolin coronavirus. The preliminary findings show that the pangolin coronavirus could be a potential intermediary that can bind to the human ACE2 receptor and thus possibly infect humans. This research is ongoing in the Gamblin laboratory.
The structural and functional analysis conducted by Dr. Gamblin's team can also potentially contribute to the development of a vaccine for SARS-CoV-2. Vaccines work by exposing the body to inactivated viruses or viral fragments (such as, for example, spike proteins) to produce an immune response without causing disease. The idea is if you are exposed to the virus in the future, your body will be prepared and able to fight the infection.
Dr. Wrobel emphasizes this research is not the direct basis for the development of a vaccine. Rather, the results of this study, in providing a detailed structural analysis of the SARS-CoV-2 spike protein and how it binds the ACE2 human receptor, may help vaccine developers optimize vaccine design.
Dr. Antoni Wrobel is a post-doctoral fellow in the Structural Biology of Disease Processes Laboratory at the Francis Crick Institute in the United Kingdom. He is a biochemist and biophysicist who uses technologies including cryogenic electron microscopy to understand the structure and function of viral proteins like those found on influenza and coronaviruses. In his free time, Dr. Wrobel likes to pick mushrooms, dance polkas, enjoy primitive art especially (but not only) from Eastern Europe, and relax with a good book from Sir Richard Burton.
Dr. Donald Benton is post-doctoral fellow in the Structural Biology of Disease Processes Laboratory at the Francis Crick Institute in the United Kingdom. He is a virologist and structural biologist who investigates the mechanisms by which viruses enter cells by using a range of techniques including electron cryo-microscopy and biophysics. In his free time, Dr. Benton likes to play the
ophicleide,
make taxidermy fish, and relax with his pet gerbil.
Dr. Steve Gamblin is the Director of Scientific Platforms in the Structural Biology of Disease Processes laboratory at the Francis Crick Institute in the United Kingdom. His laboratory focuses on determining the structure and function of proteins involved in disease. In his free time, Dr. Gamblin has a passion for antique tractors and enjoys farming crops, especially potatoes.
For More Information:
- Wrobel, A. et al. 2020. “SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects.” Nature Structural and Molecular Biology, 27: 763-7. https://www.nature.com/articles/s41594-020-0468-7
- Benton, D. et al. 2020. “Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion.” Nature. https://www.nature.com/articles/s41586-020-2772-0
- Wrobel, A. et al. 2020. “Antibody-mediated disruption of the SARS-CoV-2 spike glycoprotein.” Nature Communications, 11(5337). https://www.nature.com/articles/s41467-020-19146-5
- Wrobel, A. et al. 2020. “Structure and binding properties of Pangolin-CoV Spike glycoprotein inform the evolution of SARS-CoV-2.” Preliminary findings, available https://www.researchsquare.com/article/rs-83072/v1.
To Learn More:
General
- Structural Biology of Disease Processes Laboratory. https://www.crick.ac.uk/research/labs/steve-gamblin
- The Francis Crick Institute. https://www.crick.ac.uk/
SARS-CoV-2 and COVID-19
- World Health Organization. https://www.who.int/health-topics/coronavirus#tab=tab_1
- Centers for Disease Control and Prevention. https://www.cdc.gov/coronavirus/2019-ncov/index.html
- United Nations. https://www.un.org/en/coronavirus
Coronaviruses
- National Institutes of Health. https://www.niaid.nih.gov/diseases-conditions/coronaviruses
- Centers for Disease Control and Prevention. https://www.cdc.gov/coronavirus/types.html
Written by Rebecca Kranz with Andrea Gwosdow, PhD at www.gwosdow.com
HOME | ABOUT | ARCHIVES | TEACHERS | LINKS | CONTACT
All content on this site is © Massachusetts
Society for Medical Research or others. Please read our copyright
statement — it is important. |


{kind=link}