09/11 - When the Waste Won't Leave - MSMR: What A Year!
09.11>>When the Waste Won't Leave
When the Waste Won't Leave
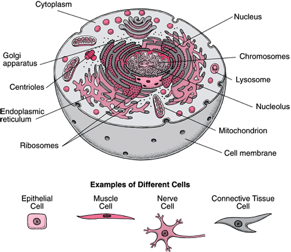
The human body is made of millions of cells with specialized functions. You can think of cells as mini factories that keep your body running: the
mitochondria produce energy for the cells, the
ribosomes produce
proteins, and the
nucleus makes sure the cell is functioning properly.
Like every factory, cells also produce waste. In order to process this waste, cells make a variety of special proteins called
enzymes to digest it, which are contained in tiny sacs called
lysosomes. These enzymes degrade cellular debris such as proteins,
fats and
carbohydrates. They can also digest many foreign substances that mistakenly come into the cell. When a cell recognizes a substance that needs to be processed, the lysosomes will
ingest it and the enzymes inside will break it down.
The body is composed of many different types of cells, each with its own structure and function. Some cells, especially glandular cells, have as their primary function the production of complex substances, such as a hormone or an enzyme. For example, some cells in the breast produce milk, some in the pancreas produce insulin. Other cells have primary functions that are not related to the production of substances−for example, muscle cells contract, allowing movement. Nerve cells generate and conduct electrical impulses, allowing communication between the central nervous system (brain and spinal cord) and the rest of the body. http://www.merckmanuals.com/home/sec01/ch001/ch001b.html
Lysosomes contain over 40 different enzymes that digest specific waste
products. If even one of these enzymes is missing or non-functional, undigested waste will begin to accumulate in the cells and can interfere with proper cell function. There is an entire class of disorders known as
lysosomal storage disorders that are characterized by the lack of a specific lysosomal enzyme. Lysosomal storage disorders can affect every system in the body, and symptoms vary greatly depending on the type of disorder and which organs are affected. Symptoms common to many lysosomal storage disorders include enlarged organs, particularly the liver and spleen, developmental delays, and impaired functioning of the nervous system. These symptoms tend to get progressively worse over time as more waste products accumulate throughout the body. Although individual lysosomal storage disorders are rare in most populations, as a group they affect approximately one in every 7,700
newborns.
Krabbe Disease
Dr. David Wenger has devoted his career to the diagnosis and treatment of lysosomal storage disorders and is the leading expert on the lysosomal storage disorder known as
Krabbe disease. Krabbe disease is a particularly debilitating disorder that affects the
nervous system, including the brain, spinal cord, and nerve cells, also known as neurons.
In general there are two forms of Krabbe disease. The infantile form, affecting about 85% to 90% of patients, appears at 3 to 4 months of age with an average age of death of 13 months. Infants presenting with this disease usually are extremely irritable and have developmental delays by about six months of age. There are also later-onset forms of the disease which affect 10% to 15% of people with Krabbe disease. The later-onset forms can present in early childhood, adolescence or even adulthood. The outcomes of the later-onset forms of the disease depend on the age the disease begins.
Patients with Krabbe disease have impaired activity of the enzyme beta-galactocerebrosidase, (shortened to GALC), which breaks down specific types of a class of molecules called
galactolipids. Galactolipids are found in high concentrations in the brain, particularly in the
myelin sheath that coats the part of the neuron known as the
axon. The axon is a projection from the nerve cell that sends electrical impulses from one cell to the next, and the myelin sheath allows them to function properly. Myelin is critical to proper neuron function because it protects axons as they send and receive messages from other neurons. The lack of myelin leads to neuronal damage and impaired neuronal function.
It is thought that the build-up of galactolipids in the brain as a result of GALC deficiency kills the cells that produce myelin, leading to the lack of myelin seen in patients with Krabbe disease.
Back to DNA and mutations
Let's start small.
Nucleotides are small molecules that join together in a particular order to form strands of
DNA. This order is very important because it provides the instructions for the creation of every protein in the human body. Like DNA, proteins are large molecules made up of strings of many smaller molecules called
amino acids. The order of amino acids in a protein is determined by the order of nucleotides in the DNA, and sets of three consecutive nucleotides encode for particular amino acids. When a series of nucleotides result in a string of amino acids that creates a protein, that segment of DNA is known as a
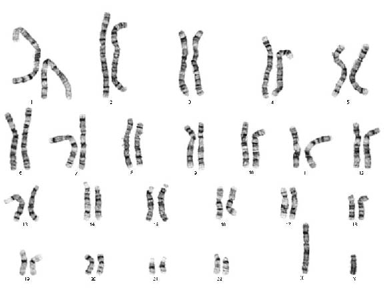
gene. In living organisms, DNA is located on
chromosomes. Every human cell contains a copy of 23 pairs of chromosomes, which comprise the entire genome. Each chromosome has a long arm and a short arm, and each arm is divided into smaller segments called bands. Each gene, therefore, has its own address within the
genome: the number of the chromosome on which it is located, the arm of the chromosome (long or short), and the band number on the arm.
Each gene also has a particular nucleotide sequence that will provide instructions for the correct amino acid sequence, resulting in the proper protein structure and function. However, during the process of cell replication there can be mistakes made in copying the nucleotide sequence. Any mistake that results in a change in the nucleotide sequence is known as a
mutation. Oftentimes mutations are harmless; in fact, they occur quite regularly. Sometimes, however, mutations can have serious consequences. If, for example, a mutation alters the amino acid sequence of a protein, the protein may be unable to function properly, becoming only partially functional or completely non-functional.
The gene mutation in Krabbe disease
In Krabbe disease, mutations in the gene that encodes for the GALC enzyme result in GALC deficiency. In 1970 researchers discovered that GALC deficiency was the cause of Krabbe disease. This made it possible for Dr. Wenger to begin studying the genetic basis of the disease. If Dr. Wenger could find the address of the GALC gene in the genome, he could make a copy of the gene and identify mutations that cause Krabbe disease. In addition, once the gene had been identified, it would be possible to begin studies for potential treatments of the disease.
To begin his search, Dr. Wenger used insight from a naturally-occurring mouse model of Krabbe disease, known as the twitcher mouse model. It lacks the same GALC enzyme as human patients. It was known that the gene for this enzyme was located on mouse chromosome 12, which corresponds to human chromosome 14. Although we now know the nucleotide sequence for the entire human genome, when Dr. Wenger was conducting this research no sequences of the GALC gene were known. In order to approximate the location of the GALC gene relative to the known sequence on chromosome 14, Dr. Wenger used a technique known as
linkage analysis. Linkage analysis calculates the probability that the disease and known sequences of other genes could be located near each other on the same chromosome given the family history of patients with Krabbe disease. Using this method and DNA from members of families where Krabbe disease was present, Dr. Wenger was able to narrow the location of the GALC gene to the long arm of chromosome 14. But he was unable to isolate the gene because there were no known sequences of DNA located near it. Instead, he had to work backwards. Rather than finding the gene directly, he had to first isolate the protein and determine the sequence of amino acids. Based on the amino acid sequence, he would then be able to derive a DNA sequence to use in linkage analysis.
Since you're going in there anyway
In order to begin this process, Dr. Wenger had to find a source of the GALC enzyme. Most organs do not produce much of this enzyme, so it was difficult to gather enough enzyme to analyze. As it turns out, urine actually contains a substantial amount of GALC. Still, Dr. Wenger needed 125 to 150 gallons of urine to obtain enough of the enzyme. So Dr. Wenger put a sign up in the men's bathroom asking people to donate their urine, and after several weeks he had enough urine to begin the process. Through many purification steps, Dr. Wenger was able to isolate the GALC enzyme. This was a complicated process, and it took over 500 liters (> 130 gallons) of urine to obtain just a few milligrams of the purified GALC enzyme.
Once the enzyme was isolated, it was then possible to identify newborns with Krabbe disease by testing for the level of enzyme in the body. Newborn genetic screening has become routine in most states in order to identify certain genetic diseases and begin any necessary treatment as soon as possible after birth. This is important in Krabbe disease and many lysosomal storage disorders because early treatment can significantly alter the outcome of the disease, reducing symptoms and slowing the progression.
In the case of Krabbe disease, the process of wrapping the axons with myelin, known as myelination, begins at about the third month of fetal development and continues rapidly into the first years of life. Nerve damage due to the lack of myelin is irreversible, and the only way to avoid this damage is to identify the disease and begin treatment as early as possible
In 2006, New York added Krabbe disease to the group of genetic disorders tested for during newborn screening. In the first four years of the program, four patients have been identified as being at-risk for the infantile form of Krabbe disease. Two of them are still alive, having lived much longer than expected as a result of successful treatment.
Bone marrow transplant, umbilical cord blood transplant
There are some possible treatment options for Krabbe disease that Dr. Wenger and other researchers are currently exploring. Even before the GALC gene was identified, researchers conducted experiments on twitcher mice to see if bone marrow transplants might increase the lifespan of the affected mice.
Bone marrow is a spongy tissue found inside bones that contains
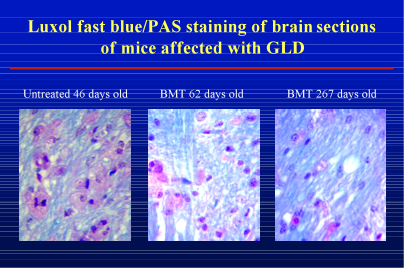
hematopoietic stem cells, the precursors of the different types of cells found in blood. In the twitcher mice, and in humans with Krabbe disease, all cells lack the GALC enzyme. Bone marrow transplants replace the defective stem cells with cells that produce normal levels of the GALC enzyme. Some of these circulating cells in the blood can get into the brain and donate some of the enzyme to the GALC-deficient cells to process the built-up waste. When researchers compared the brains of mice that received the transplant to the control mice, they observed more myelin cells and fewer white blood cells. White blood cells accumulate as a result of inflammation. This change can be seen in the following figure.
Results of bone marrow transplantation (BMT) in mice with Krabbe disease
In this image, the red stain indicates white blood cells and the blue stain indicates myelin. Untreated mice with Krabbe disease have more white blood cells (red) and less myelin (blue) when compared to treated mice. The effects of bone marrow transplantation can be seen over time as the older mice (far right panel) have even more myelin and fewer white blood cells.
These experiments indicate that bone marrow transplantation improves myelination in the brain and extends the lives of the treated mice significantly.
The best available treatment for Krabbe disease,
umbilical cord blood transplantation, is based on the same principle. Umbilical cord blood is a rich source of the same stem cells found in bone marrow, and much easier to obtain. The two patients mentioned earlier who were identified due to newborn screening are still alive thanks to umbilical cord blood transplantation. Since this treatment is so new, it is still unclear how long such a procedure can extend the life of a patient with Krabbe disease.
Researchers are also looking into a variety of other therapies. In other lysosomal storage disorders, such as
Gaucher Disease and
Fabry Disease, treatment to replace the missing enzyme, known as
enzyme replacement therapy, successfully reduces symptoms and slows the progression of the disease. In Krabbe disease, enzyme replacement therapy has proven difficult for several reasons: it is difficult to purify significant quantities of pure enzyme and it must reach the tissues needing it the most, the central and peripheral nervous systems. The blood-brain barrier is a very selective membrane that prevents large molecules such as enzymes from entering the brain cells where they are needed. A lot of research is currently focused on developing a way to deliver drugs across the blood-brain barrier.
Two other promising therapies include
gene therapy and
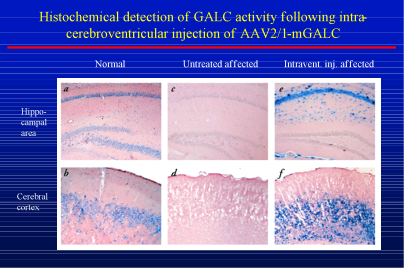
chaperone therapy. Patients with Krabbe disease have a mutation in the GALC gene that prevents it from working properly. Gene therapy uses harmless viruses to introduce a functional form of the gene to the cells needing it. This normal gene would then be able to produce normal GALC enzyme. To test whether gene therapy might be beneficial for patients with Krabbe disease, researchers injected the GALC gene into the brains of mice with Krabbe disease at two days of age. Two months later, the researchers examined two regions of the brain, the hippocampus and the cerebral cortex, to determine the level of GALC activity. The following image shows the results of these experiments.
Results of gene therapy for Krabbe disease in the mouse
In this image, the blue color shows staining for GALC activity while the pink color indicates no GALC activity. The right panels (e and f) show GALC in a treated mouse with Krabbe disease compared to the controls: an untreated normal mouse (panels a and b) and an untreated mouse with Krabbe disease (panels c and d). The blue staining of the left panels indicates normal GALC activity in the control mouse. In contrast, the absence of blue staining in the middle panels indicates the lack of GALC activity in the untreated mouse with Krabbe disease. The presence of blue staining in the right panels shows that the gene therapy has restored some GALC activity to the brain. These results are promising for future research.
Whereas gene therapy targets the gene, chaperone therapy targets the product of the gene, the enzyme itself. In many patients with Krabbe disease, the mutation in the gene for the GALC enzyme results in the production of an abnormally-shaped protein that is non-functional. Chaperone therapy would introduce small molecules that bind to the abnormal protein and stabilize it, allowing for some enzymatic activity. Both of these treatments face the same obstacles as enzyme replacement therapy, in that it is difficult to get across the blood-brain barrier. However, some small molecules do cross the blood brain barrier and viruses containing genes of interest can be injected directly into the brain.
Anti-inflammatory drugs
Recently, Dr. Wenger has conducted some experiments exploring alternative treatments to avoid the problem of the blood-brain barrier. Some of the symptoms experienced by patients with Krabbe disease are actually due to an immune response against the build−up of cellular waste. By reducing this inflammatory response, Dr. Wenger hypothesized that he could improve some of the symptoms of the disease. In these experiments, Dr. Wenger treated a mouse model of Krabbe disease that lacks a normal GALC gene with several common anti−inflammatory drugs including ibuprofen, the active ingredient in Advil. He then measured several indicators of the immune response in the brain tissues of mice who had been treated with the different drugs and compared these results to mice that had not been treated. Dr. Wenger found mice treated with the drugs had a reduced anti−inflammatory response and lived significantly longer than untreated mice. Importantly, however, when compared with mice who received a bone marrow transplant, the mice with the transplant had a much greater reduction in the immune response than any of the mice treated with anti−inflammatory drugs. "We know treatment with anti−inflammatory drugs won't cure the disease, since this is a genetic condition," Dr. Wenger remarked. "But our results show that combining anti−inflammatory drugs with other treatments might improve overall patient outcome." These are promising results indeed.
Luzi, P. et al. 2009. "Effects of treatments on inflammatory and apoptotic markers in the CNS of mice with globoid cell leukodystrophy." Brain Research, 1300: 146-158.
Duffner, P. et al. "Newborn Screening for Krabbe disease: the New York State Model." Pediatric Neurology, 40(4): 245-252.